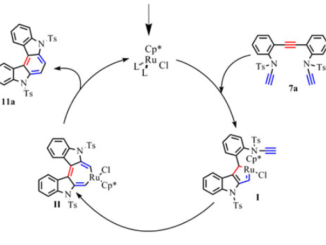
Choice of hybrid functional and basis set optimization to calculate the structural, electronic, mechanical, and vibrational properties of BaSnO3
Abstract: This work aimed to study the influence of seven hybrid functionals (B3LYP, B1WC, B3PW, PBE0, PBESOL0, SOGGAXC, and WC1LYP) and selected basis sets in the CRYSTAL software library on the calculated structural, electronic, mechanical, and vibrational properties of BaSnO3 under the framework of periodic density functional theory. Based on the results showing the best approximation of the experimental band gap energy and lattice parameter, the PBE0 functional and the Baranek basis set were used to optimize the outer valence shell functions, αsp and αd, in order to improve the quality of calculated properties. An analysis was done to determine the density of states, band structure, elastic constants, and infrared (IR) active modes. The results were compared with other reported quantum mechanical calculations and available experimental data.
Author(s): Duarte, Thiago M.; Buzolin, Prescila G. C.; Santos, Ieda M. G.; et al.
Theoretical Chemistry Accounts
Volume: 135 Issue: 6 Published: 2016
DOI: https://dx.doi.org/10.1007/s00214-016-1901-1