Abstract: The rational design of non-noble transition-metal bimetallic substrates represents a promising approach for developing efficient and tunable electrocatalysts for the hydrogen evolution reaction. In this work, we employ density functional theory calculations with van der Waals corrections combined with the computational hydrogen electrode model to investigate how hydrogen-substrate interactions govern the Gibbs free energy of hydrogen adsorption (Δ𝐺H∗) on ordered bimetallic surfaces. We investigated ordered bimetallic compounds FeNi, FeCu, and NiCu with varying atomic ratios (3:1, 1:1, and 1:3) to establish adsorption-site environment activity relationships. Our results reveal that FeNi exhibits a nearly linear dependence between Δ𝐺H∗ and the substrate ratio, showing that the catalytic activity changes by controlling the ratio of the transition-metal species. This linear scaling behavior provides a predictive framework for the rational design of experiments aimed at improving hydrogen adsorption energetics, which are governed by modulation control of the chemical species directly interfacing with the reaction environment. In contrast, although FeCu and NiCu compounds do not exhibit a linear trend as a function of specific ratios due to the local environment at the adsorption sites, the FeCu3(112) and NiCu3(111) substrates still demonstrate favorable activity. Moreover, hydrogen exhibits a strong energetic preference for hollow sites, where the adsorption energy, the dominant contribution to the Gibbs free energy, correlates directly with the chemical identity of the local catalytic site environment.
Graphical abstract:
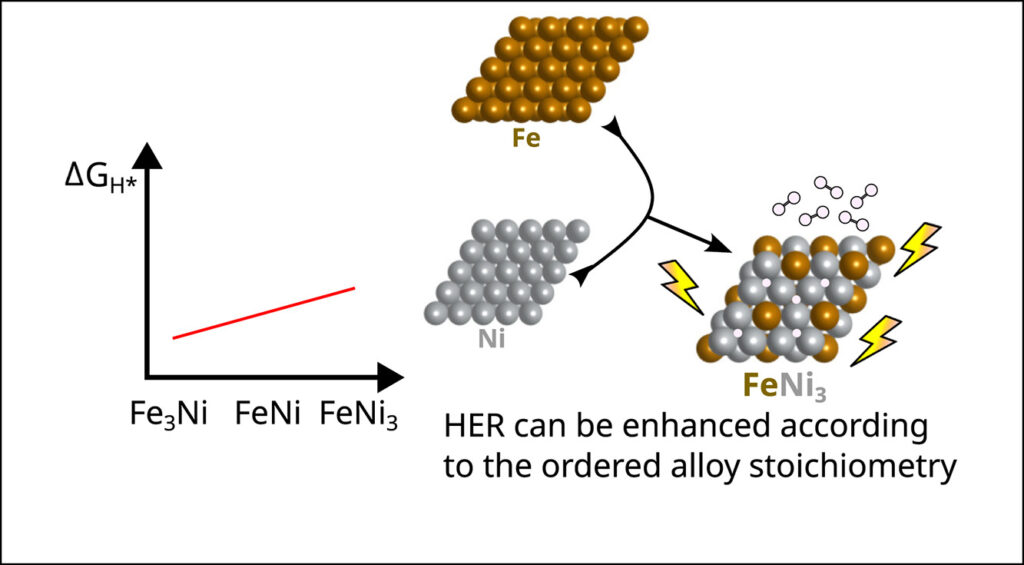
Author(s): Pedro Ivo R. Moraes, Rafael L.H. Freire, Marina Medina, Juliana F. Brito, Lucia H. Mascaro, Juarez L.F. Da Silva
First published: 25/2/2026