Writers:
Keywords: crystals; electronic structure; optical properties
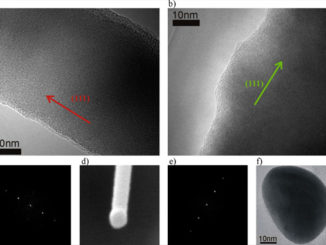
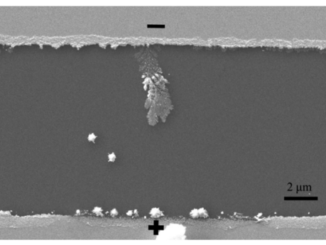
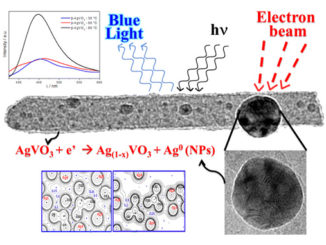
Abstract: This work combines experimental and computational techniques to exploit the best of both methodologies and thus provide a tool for understanding the geometry, cluster coordination, electronic structure, and optical properties of Ag4V2O7 crystals. This tool has been developed to create structural models and enable a much more complete and rapid refinement of 3D (three dimensional) structures from limited data, aiding the determination of structure–property relationships and providing critical input for materials simulations. Ag4V2O7 crystals were synthesized by a simple precipitation method from an aqueous solution at 30 °C for 10 min. The obtained crystals were characterized by X-ray diffraction (XRD) and Rietveld refinement, and micro-Raman spectroscopy. The crystal morphology was determined by field emission scanning electron microscopy (FESEM), and the optical properties were investigated by ultraviolet-visible (UV-vis) diffuse reflectance spectroscopy and photoluminescence (PL) measurements. The geometric and electronic structures of Ag4V2O7 crystals were investigated by first-principles quantum-mechanical calculations based on the density functional theory. The theoretical results agree with the experimental data to confirm that Ag4V2O7 crystals have an orthorhombic structure, and that the building blocks of the lattice comprise two types of V clusters, [VO4] and [VO5], and two types of Ag clusters, [AgO5] and [AgO6]. This type of fundamental studies, which combine multiple experimental methods and first-principles calculations, have been proved valuable in obtaining a basic understanding of the local structure, bonding, band gap, and electronic and optical properties of Ag4V2O7 crystals.